子昂健康: 10岁男孩身患脊髓性肌萎缩症,救命药一年75万元美金
2021-12-31 11:16:34 来源:壹点网
原来,浩浩六年前就被查出得了脊髓性肌萎缩症,这是一种以肌无力和肌肉萎缩为主要特征的常染色体隐性遗传病,不幸的是,浩浩的父母都是隐性基因携带者。
查出这种疾病之后,全家人都很害怕,因为本病目前无法治愈,1.3米高的浩浩只有50斤重,他很瘦,经常觉得很饿,但是没有办法,每天浩浩必须坚持运动,保证较好的肌肉不要萎缩。父母也在严格控制浩浩的饮食,因为他运动能力弱,如果饮食上面不控制,就可能把自己的身体给压垮,良好的饮食、生活习惯都能延缓他的疾病进程。
六年过去了,在家人、老师、同学们的照顾与帮助下,浩浩的状况还不错。不过,他的脚仍然没办法伸直,上下楼只能靠手支撑。

书法课是浩浩最困难的课程,因为这节课是在三楼的教室,其他小朋友早早就到了教室,而浩浩则需要帮忙才能上去。帮助浩浩的,是他的老师,老师扶着浩浩一步一步上楼,在耳边鼓励他加油。如果两个人困难,老师还会喊来其他的高年级的同学来帮忙。
(浩浩和楼老师 图源:钱江视频)
其他的几位任课老师也对浩浩关怀备至,不敢让浩浩独自出行,课间活动吃饭都有人陪着他。
浩浩的童年是不幸的,他没有办法像正常的孩子一样奔跑、跳跃;但是他又是幸运的,在他的身上,凝聚着家人、老师与同学的爱。
在他们所有人都在为着浩浩的健康努力时,国家也传来一个好消息,2019年2月25日,百健和昆泰联合提交的Spinraza注射液的上市申请(JXHS1800032) 获得国家药品监督管理局正式批准,用于治疗脊髓性肌萎缩症。但在国外,用这个药的费用,一年就要75万元美金,这对于一个普通患者家庭,无疑是天价药物。
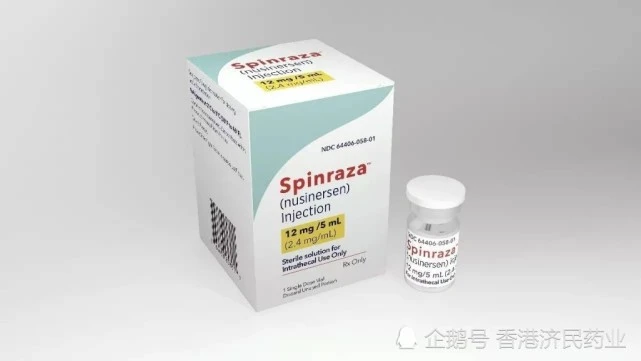
根据相关报道,目前我国国家医保局启动了动态目录调整机制,治疗大病、罕见病的药品将会逐步纳入调整的考虑范围,比如已经通过谈判纳入医保目录的诺西那生纳,这为更多罕见病患者带来了非常大的希望。
疾病科普
脊髓性肌萎缩症是一种常染色体隐性遗传病,尽管本病可由多种基因突变引起,但一般由运动神经元存活基因1 (SMN1) 突变导致。目前中国尚无发病率的确切数据。患者随着病情的进展,肌无力可进一步导致骨骼系统、呼吸系统、消化系统及其他系统异常,其中呼吸衰竭是最常见的死亡原因。
【症状】该病临床表现差异较大,根据患者起病年龄和所获得的最大运动功能,将SMA由重到轻分为4型:
脊髓性肌萎缩症I型,也称婴儿型,约占全部病例中的45%,最常见,生后6个月内发病,出现迅速发展的进行性、对称性四肢无力,最大运动能力不能达到独坐。双下肢不能抬离床面,呈髋外展、膝屈曲的蛙腿体位。患儿表情及眼球运动正常,舌肌束颤,口咽部肌群无力导致哭声低弱、吸吮无力、咽反射减弱,易发生误吸。胸式自主呼吸困难,几乎完全为腹式呼吸,吸气时腹部膨隆而胸部内陷,呈现特征性“钟形”畸形。呼吸肌无力突出,多数患儿在2岁内死于呼吸衰竭。
脊髓性肌萎缩症II型,也称中间型,约占30%-40%,生后6个月内发育正常,患儿可以获得从卧位到独坐的能力,在生后12个月左右出现四肢近端无力,上肢上举费力,始终不能独立行走,四肢感觉正常,腱反射消失。尽管寿命缩短,但多数可以活到成年期。
脊髓性肌萎缩症III型,也称青少年型,约占20%。患者在生后1年内的运动发育正常,可以获得独立行走的能力,一般在18个月后出现四肢近端无力,感觉正常,腱反射消失。随病情发展将逐渐丧失能力,预期寿命不缩短或轻度下降。
脊髓性肌萎缩症IV型,又称成人型,早期运动发育正常,多在30~60岁发病,表现为显著的四肢近端肌无力,伴随肌肉萎缩。部分患者出现小腿肌肉相对肥大,酷似肢带型肌营养不良,四肢感觉正常,腱反射消失。进展缓慢,预期寿命不缩短。
【治疗】本病目前无法治愈,预后取决于病情进展程度、治疗效果及病人体质。治疗方法有三种,分别是药物治疗、基因替代疗法与干细胞移植治疗。
药物治疗主要指反义寡核苷酸药物,上文提及的Spinraza即此类药物;还有口服药物,risdiplam;
运动神经元存活基因1基因替代疗法,通过鞘内注射方式导入体内;
干细胞移植治疗:最具潜力的是诱导型多能干细胞移植治疗。

子昂健康声明:本文作为医疗科普文章,不构成诊疗建议,不能取代医生对特定患者的个体化判断,如有就诊需要请前往正规医院。
202112201327324n4D
参考资料:
钱江视频-儿子得了罕见病脊髓性肌萎缩症,特殊药一年要百万元
爱奇艺-女童患脊髓性肌萎缩症,打一针就需70万,治疗费每年需百万
重磅喜讯! Biogen脊髓性肌萎缩症新药Spinraza在中国获批上市
子昂罕见病智库-脊髓性肌萎缩症
熊晖-《121种罕见病知识读本-脊髓性肌萎缩症》
免责声明:市场有风险,选择需谨慎!此文仅供参考,不作买卖依据。